If you use this service, could you please send us a mail to npsang@ibcp.fr with details about your usage of the NPSA service (tools used, frequency, type of sequence, ..) ?
Could you explain what makes this service unique for you ?
Could you please add information about your country and your laboratory ?
Thanks
HMMSEARCH help
A brief introduction to HMMSEARCH
The program HMMSEARCH, belonging to the HMMER package, compares a query profile HMM against a sequence library. It generates a list of significantly
similar sequence matches. This tool is very useful for homology detection, for the detection of motifs in protein sequences...
For further details, see HMMER homepage.
Parameters
The searching options correspond to the choice of the protein library. By default, the cutoff for E-values per sequence is set to 10.0 (an E-value per sequence takes into account all
matches found in the sequence) : all matches found in a sequence that passed the per-sequence threshold will be reported.
NPS@ HMMSEARCH output example
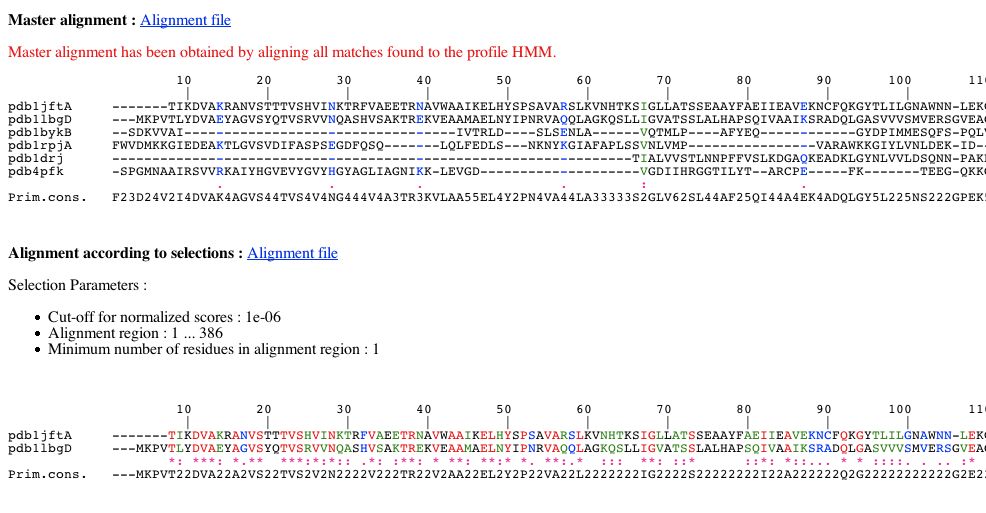
The NPS@ HMMSEARCH output is divided into four parts.
PART 1:
First, you have a form to build an alignment from matches (domains) found by HMMSEARCH. The alignment you will generate, corresponds to a
sub-alignment of the master alignment which has been obtained by aligning all matches found to the profile HMM. To build your alignment, you can :
select matches with an E-value per domain lesser or equal to a value of your choice.
By default, only matches with an E-value per domain lesser or equal to 1.0e-6 are selected.
choose an alignment region relatively to the master alignment.
Your choices are validated when you click on the REDISPLAY button.
If you want to come back to the original selections, then click on the RESET button.
PART 2:
This part corresponds to the list of matches (domains) found by HMMSEARCH. Theses matches are sorted by order of E-value (E-value per domain).
For each match, you can see :
A checkbox to select/unselect sequence.
The NPSA link allows you to apply
NPS@ methods on the corresponding sequence after it is extracted from the query database.
The database link to retrieve the database entry.
The start (seqf) and end points (seqt) of the match on the target sequence.
The E-value per sequence : this E-value takes into account all matches found in the sequence.
The domain score and the E-value per domain.
The number of displayed sequences.
The number of selected sequences.
PART 3:
You can see two alignments:
The master alignment obtained by aligning all matches found to the profile HMM. Consequently this alignment can present columns with only gaps.
The alignment according to your selections. As said previously, it corresponds to a sub-alignment of the master alignment :
this alignment is composed from matches whose E-value per domain is lesser or equal to the threshold chosen previously.
this alignment corresponds to a particular region of the master alignment according to boundaries chosen in PART 1.
PART 4:
You have :
A HMMBUILD form to follow your analysis by using HMMBUILD. The alignment which will be used
by HMMBUILD corresponds to your last selections. Columns containing only gaps will be removed. So be careful about the consistency of the generated alignment.
A checkbox allows you to remove identical sequences in the created alignment.
An EXTRACT form to extract selected sequences and make a database. You can extract full or partial sequences.
The extraction is done on your current selection.
The full extract is made from the database.
The partial extract is made from selected sequences which contain a given number of residues in the indicated alignment region.
The extracted sequences are retrieved from the HMMSEARCH alignment.
A checkbox allows you to remove identical sequences in the created database.
A link on the original HMMSEARCH result text file.

 A brief introduction to HMMSEARCH
A brief introduction to HMMSEARCH