SOPMA help
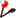 A brief introduction to SOPMA
A brief introduction to SOPMA
SOPMA (Self-Optimized Prediction Method with Alignment) is an improvement of
SOPM method. These methods are based on the homologue method
of Levin et al.. The improvent takes place in the fact that SOPMA takes into account information from an alignment of sequences
belonging to the same family. If there are no homologous sequences the SOPMA prediction is the SOPM one.
Warning : It can take up to 5 minutes to compute SOPMA for a sequence (45 seconds for RBTR_KLEAE (270 aa) and 4'33 minutes for
MDR3_HUMAN (1270 aa) ). So, be careful when using it in alignment (the total computing time can't be
above 2 hours on NPS@).
NPS@ is the orginal server for this method.
Availability in NPS@
This method is available :
Parameters
You can set the number of conformational states to predict : 3 or 4.
The similarity threshold parameter is the threshold below which a subject peptide is rejected when it's compared with a query
peptide of the sequence.
The window size parameter sets the length of the peptides to use.
NPS@ SOPMA output example
You can see:
- MPSA/ANTHEPROT link to
view the prediction in these local protein sequence analysis
softwares.
- The color coded prediction (a sequence line and below the corresponding predicted states).
- The sequence length.
- The percentage of each secondary element.
- Two graphics. The first to better visualize the prediction. In the second, there are the score curves for each predicted
state.
- The paramaters used.
- Links on the prediction result text file and on intermediate result text files.
- Links on intermediate result files to view them in MPSA/ANTHEPROT.
References