 A brief introduction to MULTALIN
A brief introduction to MULTALINMULTALIN is a progressive multiple sequence alignment program which allows you to align several biological sequences using hierarchical clustering.
For further details, see MULTALIN documentation.
A brief introduction to MULTALIN
MULTALIN is a progressive multiple sequence alignment program which allows you to align several biological sequences using hierarchical
clustering.
For further details, see MULTALIN documentation.
Availability in NPS@
MULTALIN is available :
Parameters
With the output order parameter you can choose to show the aligned sequences in the input order or in the aligned order.
The symbol comparison table sets the matrix to use scoring matches in the alignment.
The gap penalty value and the gap length value set the penalties for gaps. The alignment score is then the sum of
matches minus gap penalty value times the number of gaps and minus the gap length value times the total length of gaps.
If the extremity gap penalty option is selected, end gaps are penalized as internal gaps.
With one iteration only the alignment is done more quickly but it may not be the most accurate.
The unweighted sequences option gives the same weight (1.0) to all sequences.
The scoring method option sets how the pairwise scores are computed.
Then you set two conservation levels. For each position, MULTALIN computes the most present residue. If the residue is present
with a percentage equal or greater than the first conservation level, it will be write in upper-case letter in the consensus line. If
it's present with a percentage equal or greater than the second consensus level but lesser than the first one, it will be printed in
lower-case letter.
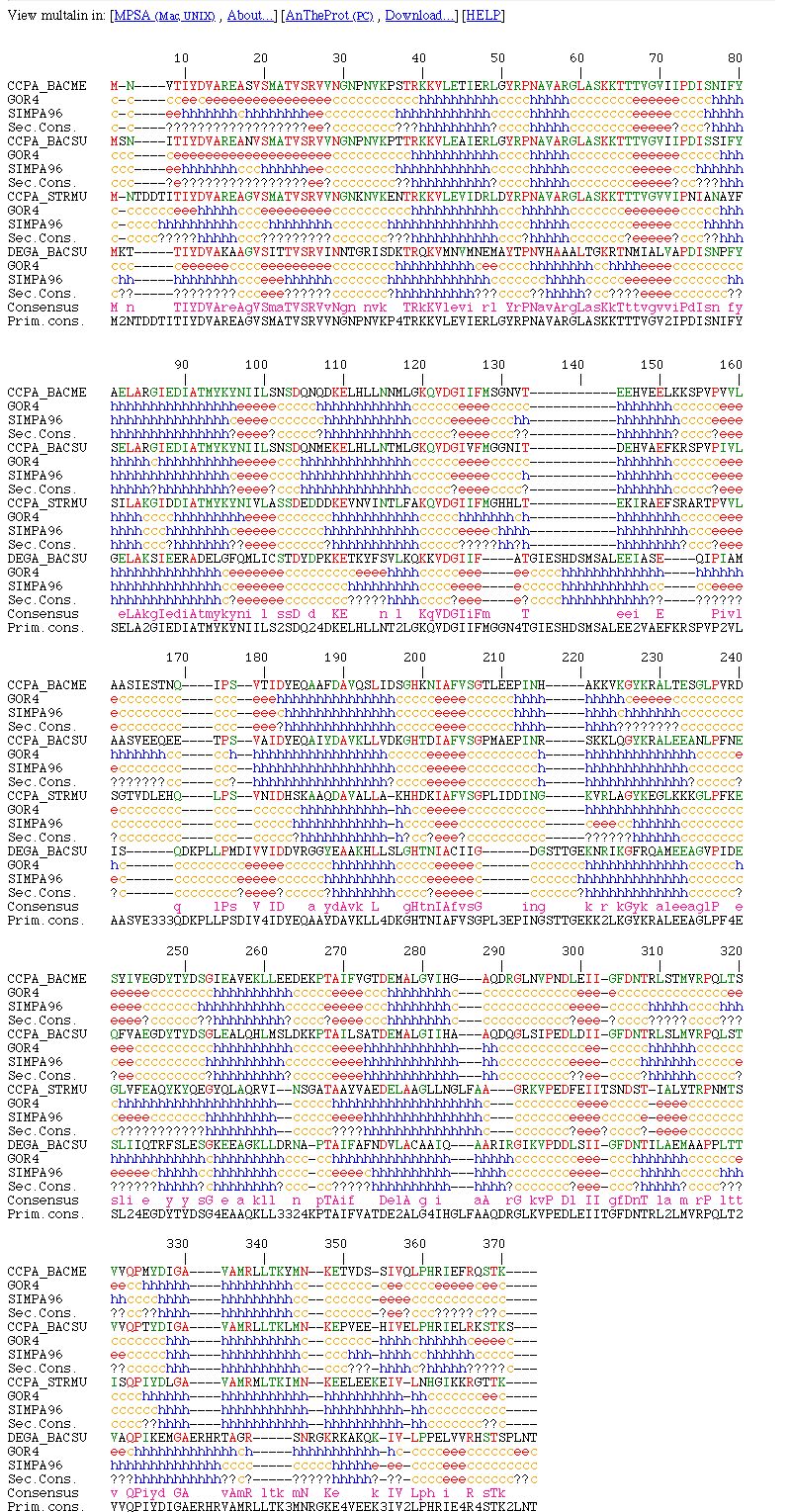
NPS@ MULTALIN output example
The NPS@ MULTALIN output is divided into three parts.