CLUSTALW help
 A brief introduction to CLUSTALW
A brief introduction to CLUSTALW
CLUSTALW is a progressive multiple sequence alignment program. It proceeds in three steps. In
the first one, all sequences are aligned by pair. Then, in the second, a dendogram is constructed describing the approximate groupings
of the sequences by similarity. In the third, and last, the multiple alignment is built using the dendogram as a guide. It takes into
account sequence weighting, positions-specific gap penalties and different weight matrix.
For further details, see CLUSTALW documentation.
Availability in NPS@
CLUSTALW is available :
The database to align cannot have more than 250,000 characters.
So, for example, you can use CLUSTALW to align a database of similar sequences built from a
BLAST search or a
PATTINPROT search.
Parameters
Five output formats are available in CLUSTALW. Only clustalw format gives the output as described below. With other formats, the
file is written to the page.
With the output order parameter you can choose to show the aligned sequences in the input order or in the aligned order
(the dendogram order).
You have to choose between fast or slow pairwise alignment. For fast pairwise alignement, you can
set the K-tuple (decrease for sensitivity), the number of tops diagonals (decrease for speed, increase for sensitivity),
the window size (decrease for speed, increase for sensitivity), the gap penalty and the scoring method. For
slow pairwise alignement, you can set the protein/DNA weight matrix, the gap opening penalty and the gap
extension penalty.
The you can set the multiple alignment parameters : the protein/DNA weight matrix, the gap opening penalty,
the gap extension penalty, the percent of identity for delay, the gap separation distance and the
no end gap separation penalty. In case of proteins, other parameters are available : the residue-specific gap penalties,
the hydrophilic residues and the hydrophilic gaps.
Pairwise alignment parameters control the speed and the sensitivity of the initial alignements.
Multiple alignment parameters control the gaps in the final multiple alignments.
NPS@ CLUSTALW output example
The NPS@ CLUSTALW output is divided into three parts.
-
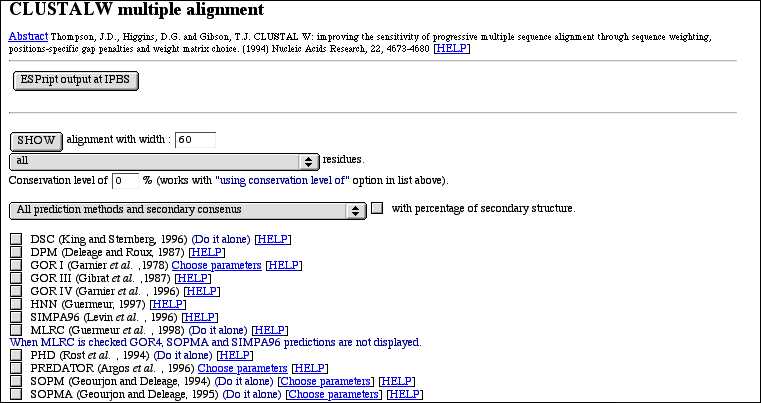
PART 1:
After the reference, you have buttons to post-process the multiple alignment in other software like ESPript,...
In this part, you have also a form to work with the alignment. Your choices are validate when you click on the SHOW button. You can :
- show residues with different options (identical(*), strongly similar (:), weakly similar (.), ...). The most
interesting option is the "using conservation level" one. With it you, display only resiues that have a conservation level
equal or above the value you type in.
- select secondary structure prediction methods to compute and display. This in the case of proteins and when you have no
more than 50 sequences. A method with the "(Do it alone)" sentence has to be computed alone (it must be
the only one newly selected before the next click on "SHOW" button). Otherwise, you won't have your response because of the
timeout (these methods can take 5 minutes or more by sequence). You can also show all methods with or without
secondary consensus or only the consensus. You can
also see the percentage of each secondary structure element (HETC...) for each method.
-
PART 2:
It's the color coded alignment with or without secondary structure predcitions inserted.
You can see :
- MPSA/ANTHEPROT link to
view data in these local protein sequence analysis softwares.
You can then download the alignment in MPSA/ANTHEPROT. With MPSA, secondary structures are downloaded too. The alignment width must be
equal to 60 for download in these softwares.
- The alignment with inserted secondary structure predcitions if any.

-
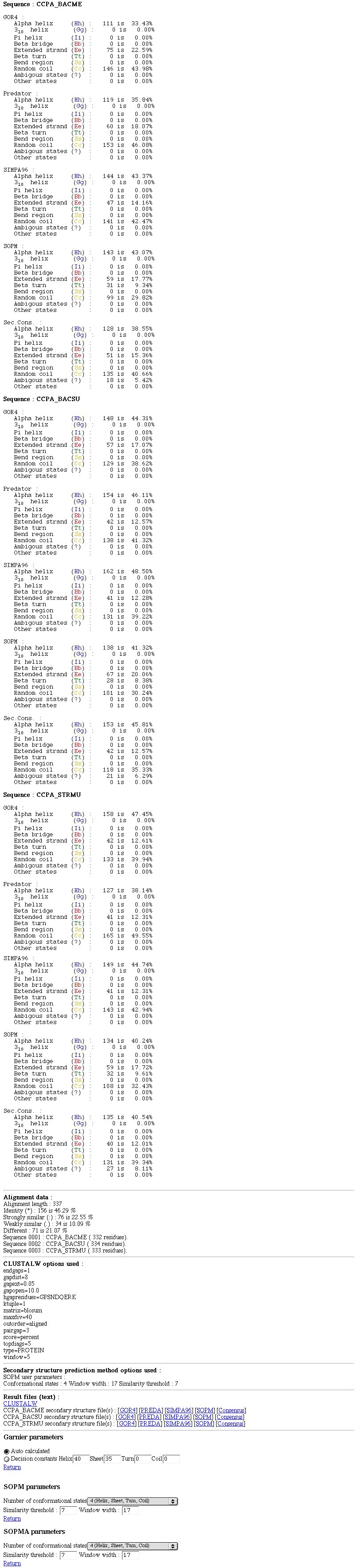
PART 3:
You have :
- The percentage of each secondary structure element (if wanted) for each prediction and for each sequence.
- Some data on the alignment (length, number of identities,...).
- CLUSTALW options used.
- Links on result text files (CLUSTALW, secondary structure prediction method outputs,...).
- Secondary struture prediction method parameters choice.
References